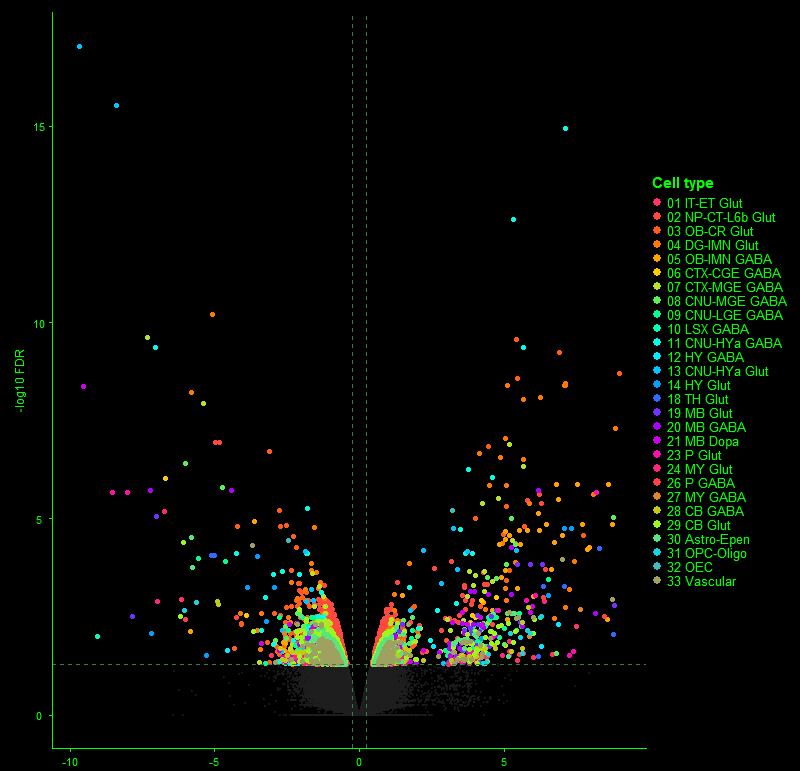
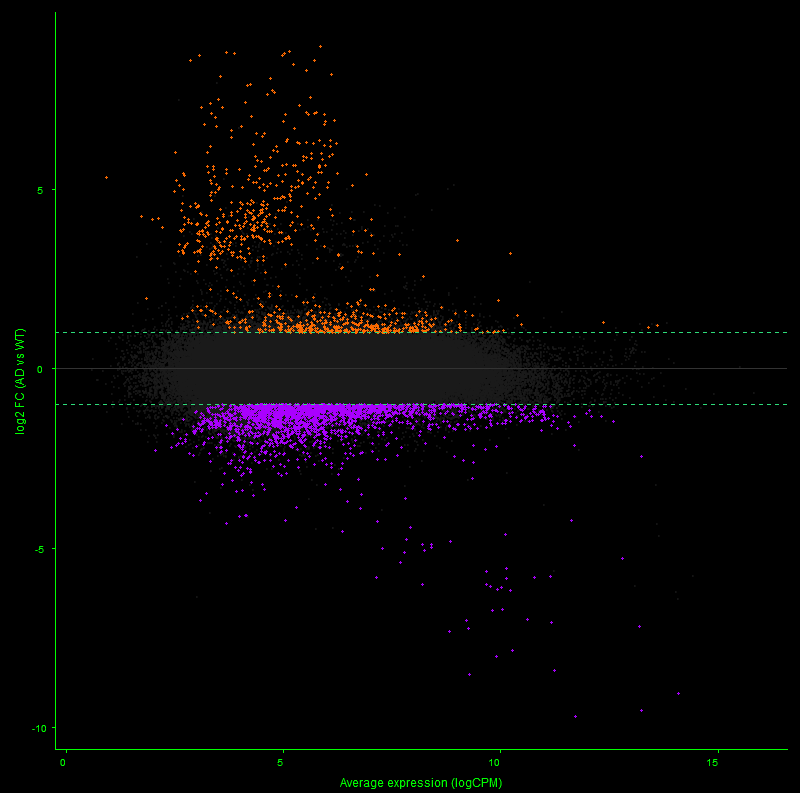
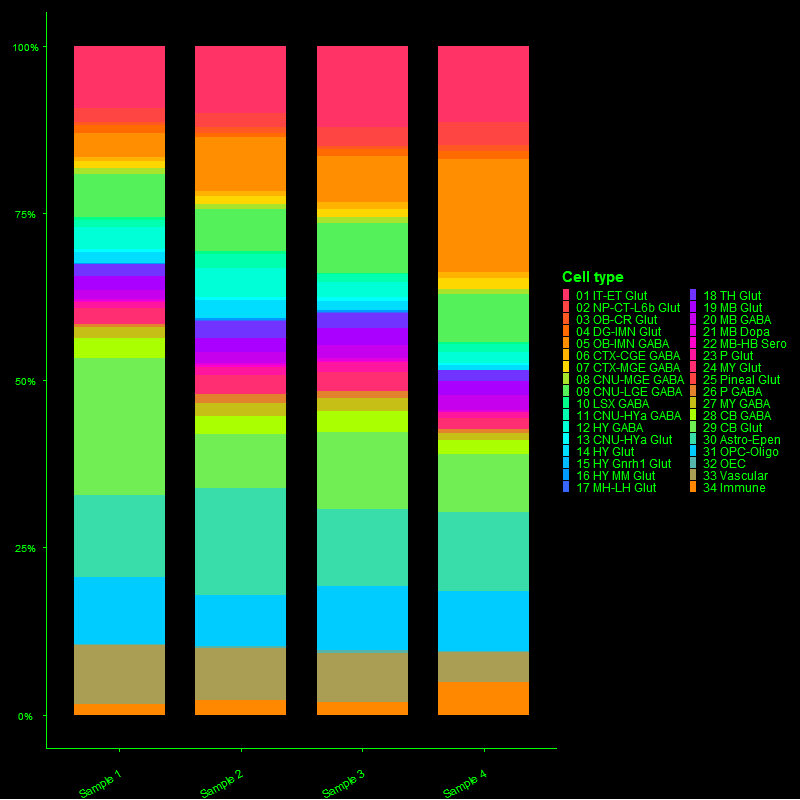
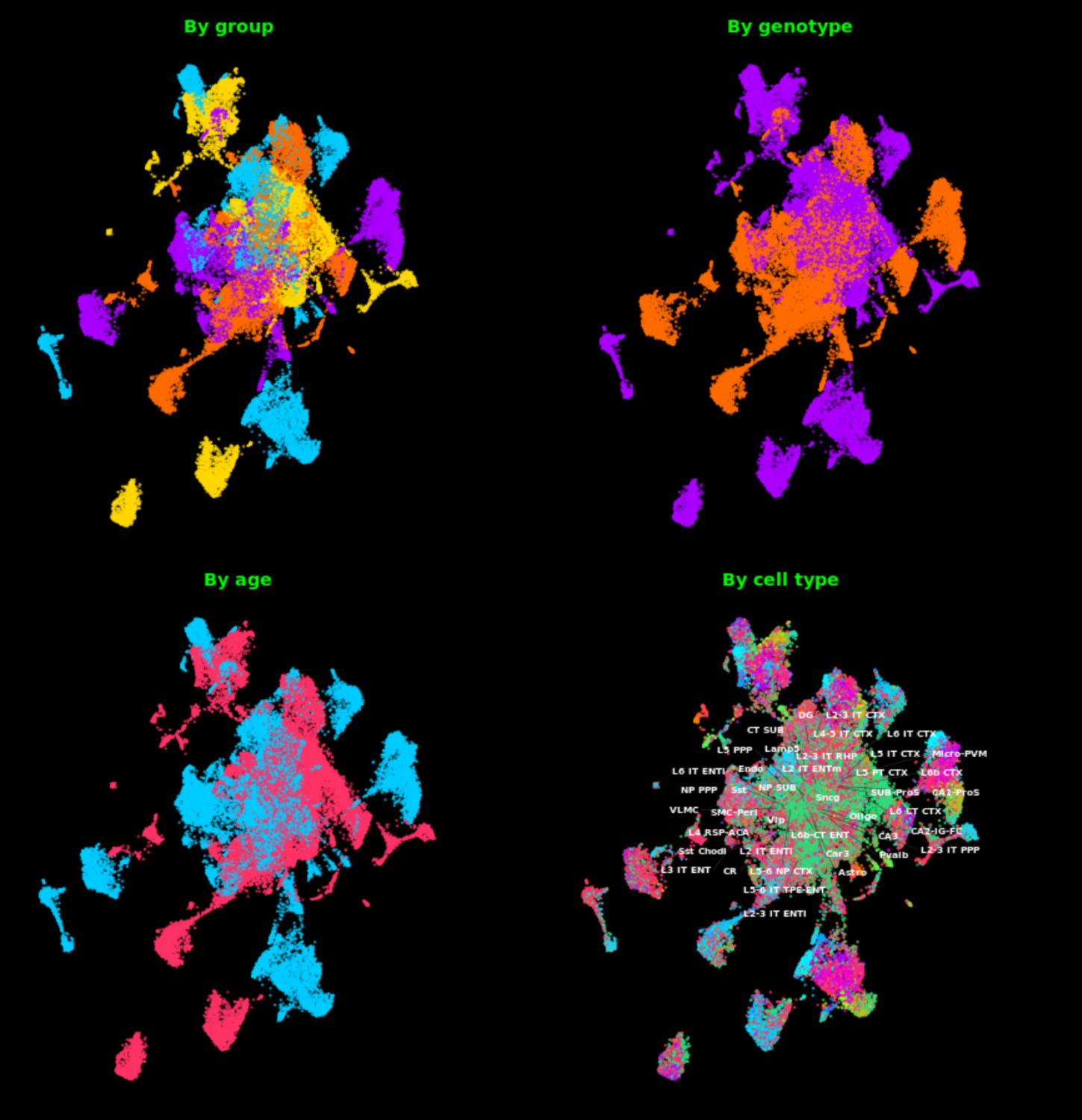
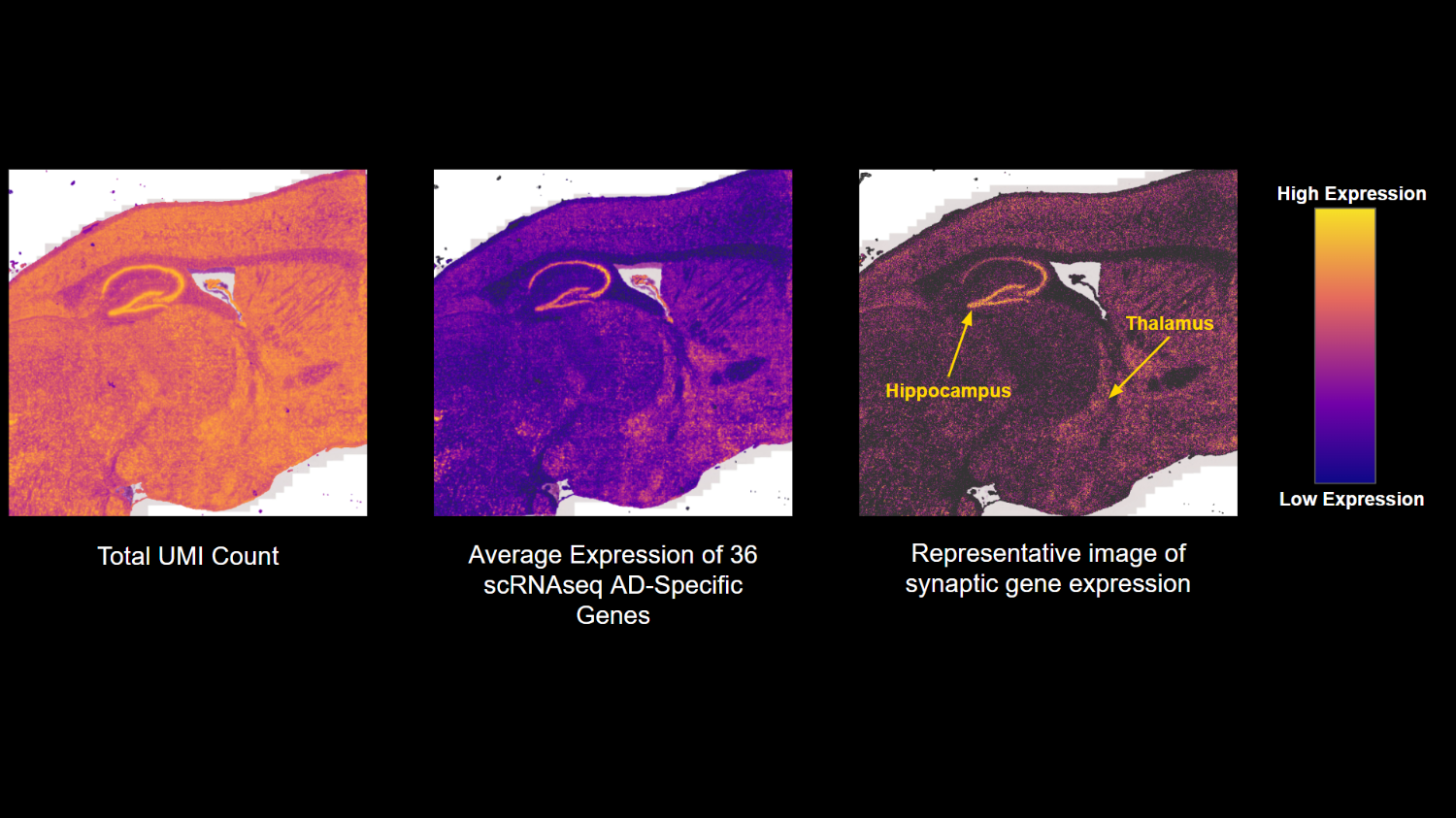
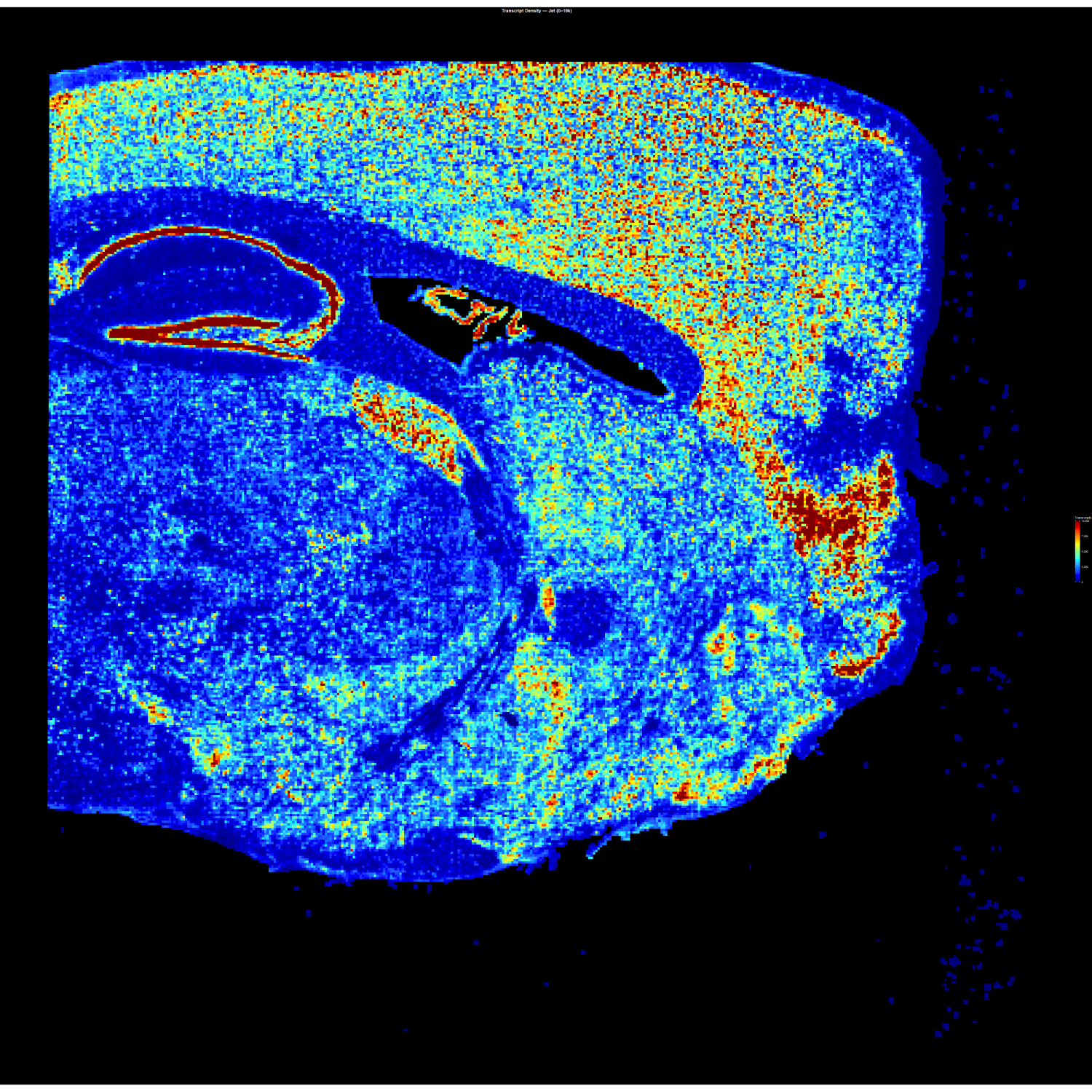
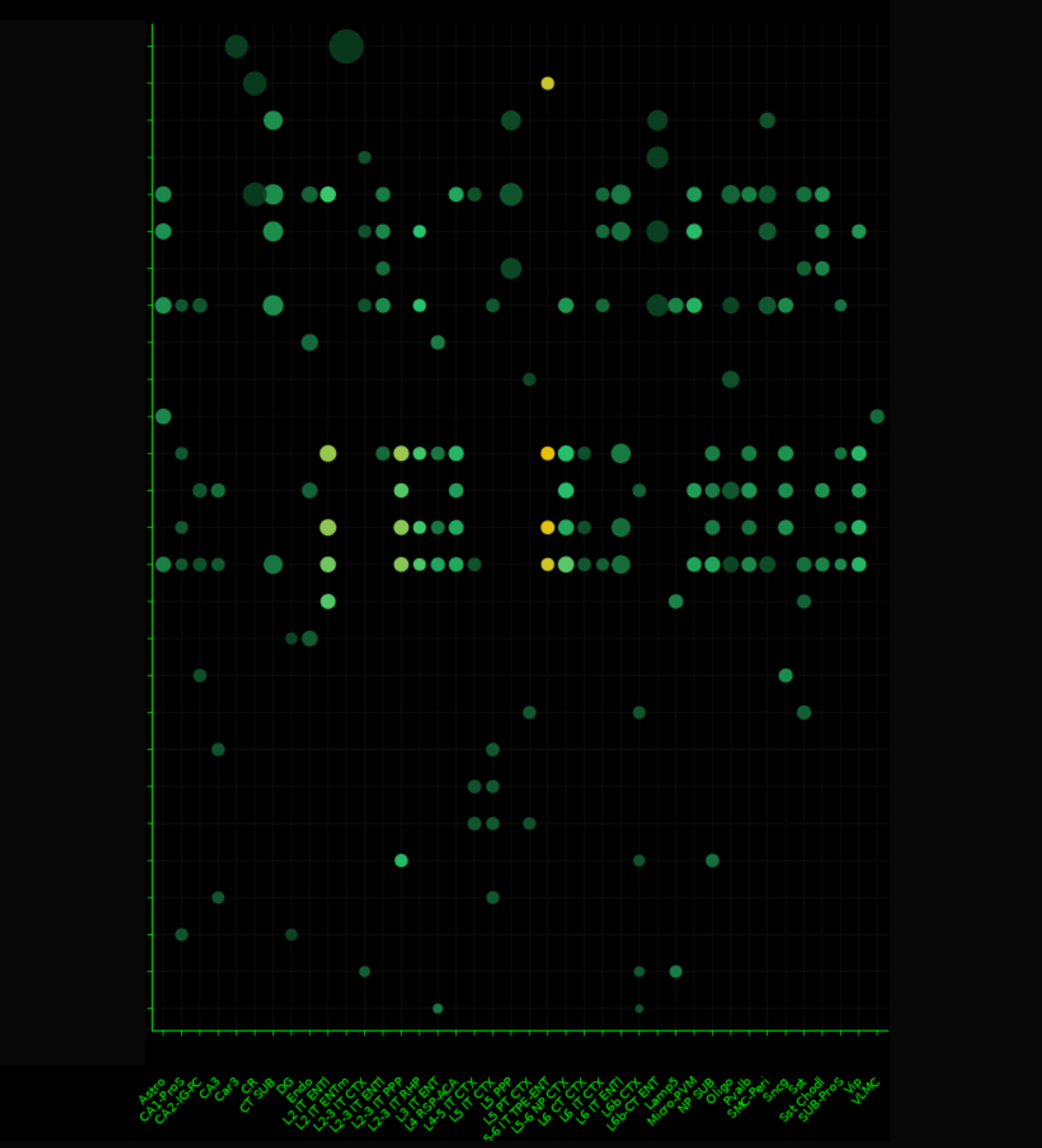
// 01 — library_prep.workflow
Library Preparation Workflows
// Parallel single-cell and spatial pipelines, from probe hybridization to sequencing.
single_cell + spatial library prepfig01.png
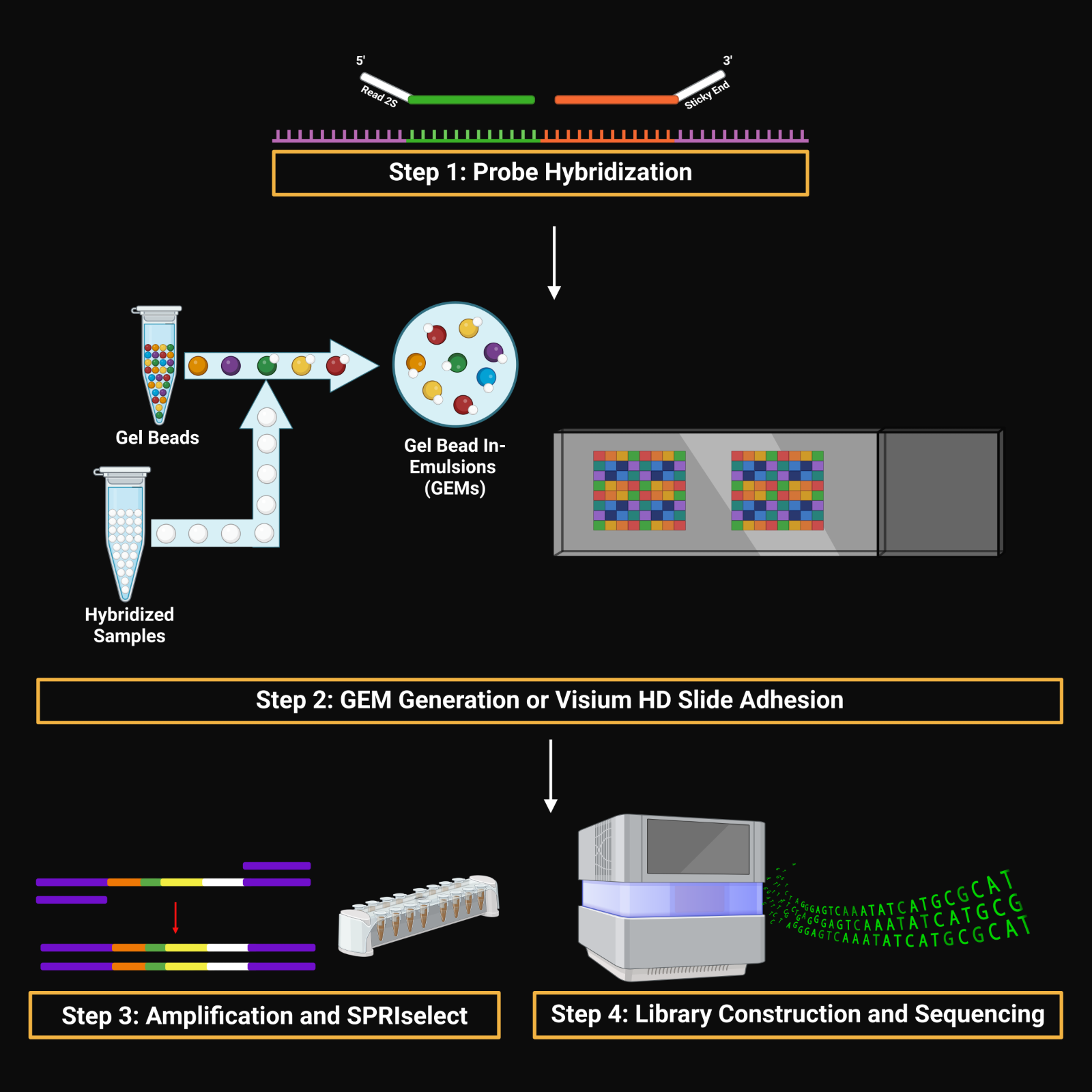
// Figure 1 — Overview of single-cell (Chromium X Fixed RNA Profiling) and spatial (Visium HD) transcriptomics library preparation workflows. Both workflows enable full transcriptome analysis at single-cell resolution, with Visium HD spatial mapping reaching 2µm resolution.